自分のデータを使用する¶
Genomon以外のデータを使用するにはconfigファイルを編集して自分のファイルフォーマットを指定します。
configファイルのサンプルは以下にあります。
{paplotをインストールしたディレクトリ}/example/example.cfg
Genomonデータを使用する場合は各バージョンの設定ファイルを用意していますので、 Genomon データを使用する 参照してください。
警告
作成したconfigファイルは pa_plot コマンドの --config_file オプションで指定します。
実行例
pa_plot qc "example/qc/*.csv" ./tmp DUMMY --config_file example/example.cfg
1. 全般¶
1 2 3 4 5 6 7 8 | ###################### general
[style]
# グラフのレイアウトファイル
# ~/tmp/paplot/style/rainbow.js
path =
# index.html の備考欄に出力するテキスト(HTMLタブ使用可, 半角英数字のみ)
remarks =
|
2. QC¶
QCグラフ固有の設定記載方法について、詳細は Config 記述方法(QC) に記載しています。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 | ###################### qc
[qc]
# (none)
# 入力フォーマット (自分のデータに合わせて変更する)
# 項目は欄外「ファイルフォーマット」参照
[result_format_qc]
suffix = .qc.csv
sept = ,
header = True
comment = #
# column index (required)
# (none)
# column index (option)
col_opt_duplicate_reads = duplicate_reads
col_opt_mapped_reads = mapped_reads
col_opt_total_reads = total_reads
col_opt_average_depth = average_depth
col_opt_mean_insert_size = mean_insert_size
col_opt_ratio_2x = 2x_rt
col_opt_ratio_10x = 10x_rt
col_opt_ratio_20x = 20x_rt
col_opt_ratio_30x = 30x_rt
col_opt_read_length_r1 = read_length_r1
col_opt_read_length_r2 = read_length_r2
col_opt_id = file_name
# 出力フォーマット (data_qc.csv)
# 項目は欄外「ファイルフォーマット」参照
[merge_format_qc]
lack_column_complement = NA
sept = ,
# 領域選択用のグラフ設定
[qc_chart_brush]
title =
title_y =
stack = {average_depth}
name_set = average:#E3E5E9
tooltip_format =
# グラフ設定(グラフごとに用意する)
[qc_chart_1]
title = depth coverage
title_y = coverage
stack1 = {ratio_30x}
stack2 = {ratio_20x-ratio_30x}
stack3 = {ratio_10x-ratio_20x}
stack4 = {ratio_2x-ratio_10x}
name_set = ratio_30x:#2478B4, ratio_20x:#FF7F0E, ratio_10x:#2CA02C, ratio_2x:#D62728
tooltip_format1 = ID:{id}
tooltip_format2 = ratio_2x: {ratio_2x:.2}
tooltip_format3 = ratio_10x: {ratio_10x:.2}
tooltip_format4 = ratio_20x: {ratio_20x:.2}
tooltip_format5 = ratio_30x: {ratio_30x:.2}
|
3. CA¶
CAグラフ固有の設定記載方法について、詳細は Config 記述方法(CA) に記載しています。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 | ###################### sv
[genome]
# ゲノムサイズのファイル(CSV形式)(デフォルトはhg19, installディレクトリ配下のgenomeディレクトリにあります)
#
# for example.
# (linux)
# path = ~/tmp/genome/hg19.csv
# (windows)
# path = C:\genome\hg19_part.csv
path =
[sv]
# 使用するchromosomes (,で区切る)
use_chrs = 1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,X,Y
# if setting label-text & color
# use_chrs = 1:Chr1:crimson, 2:Chr2:lightpink, 3:Chr3:mediumvioletred, 4:Chr4:violet, 5:Chr5:darkmagenta, 6:Chr6:mediumpurple
# 積み上げグラフのchromosome分割サイズ (bps)
selector_split_size = 5000000
# 入力されていた場合、そのgroupのみ出力する
# 未入力の場合、検出されたgroupすべて出力する
# , 区切りで複数指定可能
#
limited_group = stopgain,frameshift_deletion,frameshift_insertion
# 入力されていた場合、そのgroupはplot対象から除外する
# , 区切りで複数指定可能
# 空白行を除去する場合、_blank_ と記入する
nouse_group = _blank_,unknown,synonymous_SNV
# groupのplot色を指定する。group名:(RGBもしくはカラー名)
# , 区切りで複数指定可能
# 未入力のgroupはデフォルト色を使用する
group_colors = stopgain:#E85299,frameshift_deletion:#F39600,frameshift_insertion:#E60011
# 入力フォーマット (自分のデータに合わせて変更する)
# 項目は欄外「ファイルフォーマット」参照
[result_format_sv]
suffix = .result.txt
sept = \t
header = False
comment = #
# column index (required)
col_chr1 = Chr_1
col_break1 = Pos_1
col_chr2 = Chr_2
col_break2 = Pos_2
# column index (option)
col_opt_dir1 = Dir_1
col_opt_dir2 = Dir_2
col_opt_type = Variant_Type
col_opt_gene_name1 = Gene_1
col_opt_gene_name2 = Gene_2
col_opt_group =
col_opt_id =
# 出力フォーマット (data_sv.csv)
# 項目は欄外「ファイルフォーマット」参照
[merge_format_sv]
lack_column_complement = NA
sept = ,
|
4. mutation-matrix¶
mutation-matrixグラフ固有の設定記載方法について、詳細は Config 記述方法(mutation-matrix) に記載しています。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 | ###################### mutation
[mut]
# geneのサンプルに対する検出比(%)
# 値より小さいgeneはplot対象から除外する
# 0の場合はすべて出力する
use_gene_rate = 0
# 入力されていた場合、そのgeneのみ出力する
# 未入力の場合、検出されたgeneすべて出力する
# , 区切りで複数指定可能
#
# limited_genes = TP,TTN,APC,BRAF,CDH1,FLT3
limited_genes =
# 入力されていた場合、そのgeneはplot対象から除外する
# , 区切りで複数指定可能
#
# nouse_genes = NONE,MUC4
nouse_genes =
# 入力されていた場合、その変異タイプ(func)のみ出力する
# 未入力の場合、検出されたfuncすべて出力する
# , 区切りで複数指定可能
#
# limited_funcs = exome,splicing
limited_funcs =
# 入力されていた場合、そのfuncはplot対象から除外する
# , 区切りで複数指定可能
# 空白行を除去する場合、_blank_ と記入する
nouse_funcs = _blank_,unknown,synonymous_SNV
# funcのplot色を指定する。func名:(RGBもしくはカラー名)
# , 区切りで複数指定可能
# 未入力のfuncはデフォルト色を使用する
func_colors = stopgain:#E85299,frameshift_deletion:#F39600,frameshift_insertion:#E60011,nonframeshift_deletion:#9CAEB7
# ポップアップウィンドウの表示内容
# 詳細は以下
tooltip_format_checker_title1 = ID:{id}, gene:{gene}, {#sum_item_value}
tooltip_format_checker_partial = type[{func}], {chr}:{start}:{end}, [{ref} -----> {alt}]
tooltip_format_gene_title = gene:{gene}, {#sum_item_value}
tooltip_format_gene_partial = func:{func}, {#item_value}
tooltip_format_id_title = ID:{id}, {#sum_item_value}
tooltip_format_id_partial = func:{func}, {#item_value}
# 入力フォーマット (自分のデータに合わせて変更する)
# 項目は欄外「ファイルフォーマット」参照
[result_format_mutation]
suffix =
sept = \t
header = True
comment = #
# funcが1セルに複数入力されている場合の区切り文字
sept_func = ";"
# geneが1セルに複数入力されている場合の区切り文字
sept_gene = ";"
# column index (required)
# func列
col_func = Merge_Func
# gene列
col_gene = Gene.refGene
# column index (option)
# chromosome
col_opt_chr = Chr
# 開始位置
col_opt_start = Start
# 終了位置
col_opt_end = End
# リファレンスの塩基配列
col_opt_ref = Ref
# 対象の塩基配列
col_opt_alt = Alt
# id (sample) 列
col_opt_ID = id
# 出力フォーマット (data_mut.csv)
# 項目は欄外「ファイルフォーマット」参照
[merge_format_mutation]
lack_column_complement = NA
sept = ,
|
5. 共通項目¶
suffixとID¶
paplotではサンプル名が必要です。ファイル入力では、以下のことに注意してください。
case1: マージされたファイルを入力する
複数サンプルの結果が、1ファイルにすべてまとめられていると想定しています。サンプル名となる列を
col_opt_IDで必ず指定してください。case2: サンプルごとに分かれた複数のファイルを入力し、データ中にサンプル名となるものはない。
ファイル名の一部をサンプル名として使用します。
suffixを必ず指定してください。case3: サンプルごとに分かれた複数のファイルを入力し、データ中にサンプル名となるデータがある。
サンプル名となる列を
col_opt_IDで必ず指定してください。
複数ファイル入力する場合のコマンドの実行方法は pa_plot コマンド を参照してください。
入力ファイルフォーマット¶
configファイル中、[result_format_*] というセクションでは入力ファイルのフォーマットを指定します。
| suffix: | suffixとID を参照してください。 |
|---|---|
| sept: | データ区切り。 |
# タブ区切りの場合
sept = \t
# ,区切りの場合
sept = ,
# スペース区切りの場合
sept = " "
| header: | 先頭1行がヘッダかどうか。先頭行がヘッダの場合はTrue。ヘッダなしの場合はFalse |
|---|---|
| comment: | 先頭に指定文字がある行は飛ばす |
出力ファイルフォーマット¶
configファイル中、[merge_format_*] というセクションでは出力ファイル(data_*.csv) のフォーマットを指定します。
通常、変更する必要はありません。
| sept: | データ区切り。(入力ファイルフォーマットと同) |
|---|---|
| lack_column_complement: | |
| カラムがない場合、何で埋めるか | |
列の指定方法¶
ヘッダの有り無しに合わせて、カラム名もしくはカラムインデックスを入力します。
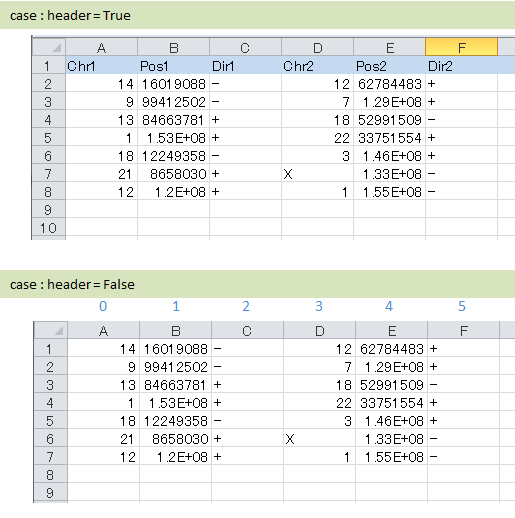
記入例
# ヘッダ行がある場合、カラム名 (テキスト) を入力する
header = True
col_chr1 = Chr_1
col_break1 = Pos_1
col_chr2 = Chr_2
col_break2 = Pos_2
# ヘッダ行がない場合、カラムインデックス (数値) を入力する
header = False
col_chr1 = 0
col_break1 = 1
col_chr2 = 3
col_break2 = 4
ユーザ定義フォーマット¶
設定例
tooltip_format_checker_partial = type[{func}], {chr}:{start}:{end}, [{ref} -----> {alt}]
表示例:
type[exome], chr1:2000:2001, [A -----> T]
col_ もしくは col_opt_ を除いた名前です。`col_opt_{任意の名前}` として追加し、実際のデータの列名を指定してください。col_opt_new_option = column_name** mutation **
| option名 | キーワード |
|---|---|
| col_func | {func} |
| col_gene | {gene} |
| col_opt_chr | {chr} |
| col_opt_start | {start} |
| col_opt_end | {end} |
| col_opt_ref | {ref} |
| col_opt_alt | {alt} |
| col_opt_id | {id} |
** ca **
| option名 | キーワード |
|---|---|
| col_chr1 | {chr1} |
| col_break1 | {break1} |
| col_chr2 | {chr2} |
| col_break2 | {break2} |
| col_opt_id | {id} |
| col_opt_dir1 | {dir1} |
| col_opt_dir2 | {dir2} |
| col_opt_type | {type} |
| col_opt_gene_name1 | {gene_name1} |
| col_opt_gene_name2 | {gene_name2} |
** qc **
| option名 | キーワード |
|---|---|
| col_opt_duplicate_reads | {duplicate_reads} |
| col_opt_mapped_reads | {mapped_reads} |
| col_opt_total_reads | {total_reads} |
| col_opt_average_depth | {average_depth} |
| col_opt_mean_insert_size | {mean_insert_size} |
| col_opt_ratio_2x | {ratio_2x} |
| col_opt_ratio_10x | {ratio_10x} |
| col_opt_ratio_20x | {ratio_20x} |
| col_opt_ratio_30x | {ratio_30x} |
| col_opt_read_length_r1 | {read_length_r1} |
| col_opt_read_length_r2 | {read_length_r2} |
| col_opt_id | {id} |
数値計算させることもできます。その場合、計算式を{}で囲います。
{#number_mutaion_gene/#number_id*100}%
表示例:
3.33333333333333%
表示桁数を指定したい場合は計算式の後に ":.2" と書きます。小数点以下3桁の場合は ":.3" と書きます。
{#number_mutaion_gene/#number_id*100:.2}%
表示例:
3.33%